





百濟神州PD-1“出海”延期!中國Biotech國際化路徑再考量
2022年是中國醫藥創新國際化的大年。
隨著信達生物信迪利單抗、君實生物特瑞普利單抗陸續折戟,去年9月百濟神州向美國食品藥品監督管理局(FDA)提交的PD-1抗體百澤安(替雷利珠單抗注射液)上市申請結果,備受行業關注。
就在剛剛跨過PDUFA日期2022年7月12日之際,7月14日晚間,百濟神州發布公告,披露替雷利珠單抗注射液的申報進展。
公告顯示,美國FDA因新冠肺炎疫情相關的旅行限制,無法如期在中國完成所需的現場核查工作,因此將延長替雷利珠單抗針對不可切除或轉移性食管鱗狀細胞癌(ESCC)患者的二線(2L)治療的新藥上市許可申請的審評時間,直至現場核查完成。
國產創新藥出海道路曲折。前有百濟神州BTK抑制劑澤布替尼膠囊敲開美國FDA大門,取得中國小分子創新藥美國獲批“零的突破”,并陸續在世界市場50個國家和地區獲批上市;后有大分子抗體藥物信達生物、君實生物等企業闖關沖刺,卻沒能取得預期的成果;再有細胞治療CAR-T產品傳奇生物一舉奪魁。中國創新創新藥國際化前景依然令人振奮。
近期,君實生物已經向美國FDA重新提交了特瑞普利單抗上市申請,百濟神州替雷利珠單抗后續的審評進展同樣值得期待;雖然信達生物信迪利單抗在非小細胞肺癌一線治療方面遭遇坎坷,但是后續的適應癥拓展值得深耕。
誰能夠在PD-1藥物海外上市拔得頭籌,依然充滿懸念。
01、ESCC一線治療競爭?替雷利珠單抗直面考驗
近年來,中國創新藥出海動作不斷,其中既包括正在轉型的大藥企如恒瑞醫藥等,也包括醫藥新星Biotech企業如百濟神州等。蓄力闖關美國市場,不僅意味著“出海”光環,背后更意味著巨大的全球潛力市場:拿下FDA入場券,就約等于拿到了通往全球藥品銷售的“門票”。
替雷利珠單抗是百濟神州繼澤布替尼之后,最受關注的自主研發創新藥,也是公司第二款沖刺美國上市的中國新藥。
本次遭遇審評延期的適應癥ESCC,是消化道領域最常見的惡性腫瘤之一食管癌的兩種亞型之一,屬于世界范圍內食管癌最常見的亞型,占全球食管癌的85%以上,占美國食管癌病例的30%。在美國,每年食管癌的確診人數超過18400人。
由于許多患者確診時已處于疾病晚期,ESCC總體的預后較差,治療極具挑戰性,存在迫切的未被滿足的醫療需求,臨床用藥場景較為明確。
就在美國FDA宣布審評延期的半個月前,百濟神州與諾華宣布PD-1抗體替雷利珠單抗聯合化療一線治療ESCC的全球三期臨床RATIONALE 306獲得成功;相比于化療將mOS從10.6個月延長到17.2個月,延長6.6個月,降低死亡風險34%(HR=0.66)。僅從數值結果來看,可以說是目前PD-1一線治療ESCC的亮眼數據。
翻閱以往細分領域的臨床研究結果,默沙東K藥、百時美施貴寶O藥分別延長了2.6個月、2.5個月。在國產PD-1中,恒瑞醫藥卡瑞利珠、信達生物信迪利單抗、君實生物特瑞普利單抗分別將mOS延長了3.3個月、4.2個月、6.0個月。雖然相關研究沒有“頭對頭”直接對比,但數據結果依然是醫學界和產業界關注的焦點。
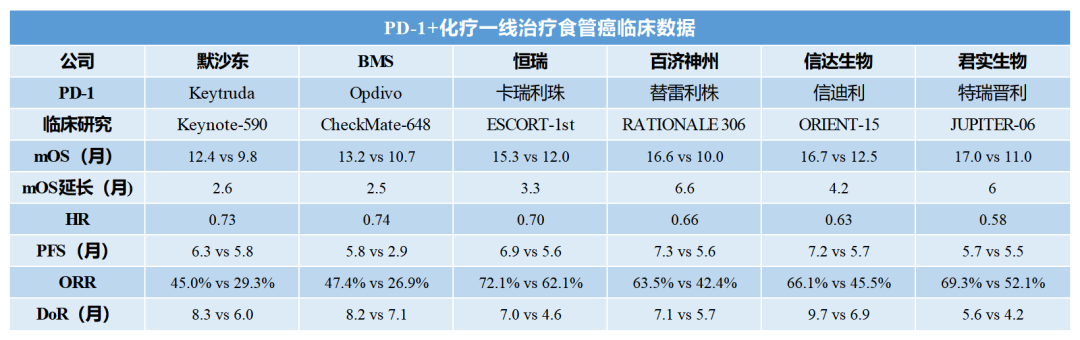
業內人士分析,替雷利珠單抗在一線治療ESCC獲得的臨床數據,將可能給此次美國審批增加更多確定性。“國產PD-1產品的國內市場價格競爭膠著,國內醫保支付大門打開,各大企業通過擴展適應癥,持續擴大競爭戰場,出海成為打破‘內卷’的重要選擇;產品如果能夠在美國獲批,這將意味著獲得高端市場的商業化競爭機會。”
與此同時,國際化產品的全球商業化挑戰,這將是每一款“出海”產品必須面對的考驗。以澤布替尼為例,作為百濟神州第一個自主研發的,也是國內第一款把國產創新抗癌藥送到美國市場上的產品,美國市場是最核心的銷售地區,自2019年11月獲FDA批準,2年內銷售額雖然持續放量增長,但是商業化表現也只是差強人意。
2020年在美銷售額為4170萬美元,2021年在美銷售額為1.16億美元(全球2.18億美元),2022年一季度在美銷售額為6405萬美元(全球銷售額為9850萬美元)。與同為BTK抑制劑的伊布替尼相比,雖然,同類競品市場爭奪激烈,依布替尼增速有所放緩,但全球銷售額依然超過97億美元。
不論是澤布替尼,還是替雷利珠單抗,亦或是未來的信迪利單抗和特瑞普利單抗,PD-1登錄全球市場,從研發到成功上市只是創新藥生命周期的“萬里長征第一步”,一個產品能否最終成為“全球重磅”,核心標準還是要看商業化成績。
顯然,無論是“出海”創新藥,還是在國內布局創新藥,任何一個相對成熟的治療領域,頭部空間都是有限的。取得競爭優勢的核心關鍵始終在于能否體現產品的創新性,滿足未被滿足的臨床需求、提高藥物可及性、改進工藝、適應癥選擇、提高安全性療效性等多個維度,都可以通過高價值創新打破“內卷”。
02、“出海”道阻且長 須對目標市場充分考量
百濟神州正在面對的全球商業化考驗,對于國內大多想要實現創新藥出海的藥企來說還為時尚早,順利實現產品“出海”依然是迫切需要解決的主要問題。
在藥品集采、醫保控費等多重政策疊加影響下,出海正成為中國醫藥企業尋求估值溢價的關鍵破局路徑,但道路充滿泥濘:
2021年12月,萬春藥業普那布林美國上市申請遇挫,FDA指出普那布林提供的注冊試驗結果不足以證明其獲益,需要進行額外的對照試驗提供支持CIN適應癥的實質性證據。
2022年2月,FDA未批準信達生物與禮來共同開發的信迪利單抗關于治療一線非鱗非小細胞肺癌的新藥上市申請。FDA強調ORIENT-11并不具有對美國人群和美國醫療實踐的普遍適用性,包括ORIENT-11的臨床試驗方案采用化療而非免疫治療,未包含OS數據;試驗沒有反映美國境內的不同種族群體等。
2022年5月,和黃醫藥發布公告稱FDA已就索凡替尼用于治療胰腺和非胰腺神經內分泌瘤的新藥上市申請發出完整回復函。FDA認為,當前基于兩項成功的中國III期研究以及一項美國橋接研究的數據包尚不足以支持藥品現時于美國獲批。
2022年5月,君實生物收到FDA關于去年3月國產PD-1特瑞普利單抗上市申請的完整回復信,要求進行一項質控流程變更。君實生物首次出海因此受阻。7月6日,FDA受理了君實生物重新提交的PD-1單抗特瑞普利單抗聯合吉西他濱/順鉑的BLA。
2022年7月,傳奇生物通知FDA,基于其一款與LB1901表達相同CAR蛋白的類似CAR-T候選產品在一項研究者發起的臨床研究中未顯示出明顯的臨床獲益,終止其針對CAR-T新藥LB1901的新藥臨床試驗申請(IND)的1期臨床試驗。美國FDA曾因該產品僅有的一名接受注射的參與者出現了細胞計數低的情況,通知傳奇生物該項臨床暫停,并將于3月11日前提供正式的臨床暫停函。
國產創新藥在美上市遇到的障礙其實非常相似。這些受挫案例也清晰表達了美國FDA的立場:要求臨床試驗及數據能充分代表美國的醫療實踐和患者人群。
事實上,從已經公開的信息可以看出,美國FDA一直非常關注藥物的創新性,在創新療法和創新藥物方面的審評審批一直都是全球醫藥創新的“風向標”。2021年,FDA藥物評價與研究中心(CDER)共批準了50個創新藥,最近五年,每年CDER批準的創新藥數量均維持在50個左右的高水平;此外,近年創新藥借助FDA加速通道審批程序獲得批準上市的比例大幅增加,2021年3/4獲批新藥使用了兩種或更多的加速通道審批程序。
專家表示,對于美國FDA而言,監管部門審評一款海外產品,關心的也是藥品本身的臨床數據能否反映真實的臨床用藥情況,包括醫療實踐、患者人群以及疾病特征等。“從美國FDA已經發布的政策、規劃,以及每年獲批的創新藥品情況,可以明顯感受到,美國FDA對于能夠滿足未被滿足臨床需求的嚴重疾病或罕見病藥物,具有療法突破和創新技術的藥物,前沿制造技術的藥物,都是非常支持和鼓勵的,這也意味著中國創新藥‘出海’需要更多聚焦臨床亟需和前沿創新。”
在萬春醫藥的普那布林、信達生物的信迪利單抗、和黃醫藥的索凡替尼、君實生物的特瑞普利單抗等國產創新藥產品接連闖關FDA受挫后,百濟神州依然保留著懸念。
據了解,百濟神州RATIONALE 306試驗的649例患者來自亞太、歐洲和北美的162家研究中心,其中57%來自中國,25%來自歐美,18%來自日韓等其他國家和地區。此外,研究者團隊充分考慮亞洲和歐美地區專家在食管癌治療臨床實踐中的差異,RATIONALE 306研究的化療方案選擇也更具有包容性。
業內人士認為,針對中國患者人群和境外患者人群的不同,要積極同步開展臨床試驗,如以國際多中心臨床研究的方式,在相同臨床試驗設計和實施條件下,對不同患者人群同步開展人體PK、PD、有效性和安全性等系統臨床試驗。
中國藥企想要在出海之路上非一帆風順,甚至在商業化上占有優勢,充分認識國際研發和我國的異同或許就是關鍵因素之一,一方面尊重目標國家當地市場的監管原則,對其市場注冊監管、商業及競爭、準入、投資及稅收環境等有全面深入的理解;另一方面,真正對目標市場的患者群體進行全盤分析。


